Help topics |
[Go back] |
RNAz [1] detects functional RNA secondary structures in multiple sequence alignment based on two characteristic features: (i) thermodynamic stability, (ii) structural conservation.
It is easy to calculate the MFE as a measure of thermodynamic stability for a sequence using e.g. RNAfold [2]. However, the MFE depends on the length and the base composition of the sequence and is, therefore, difficult to interpret in absolute terms. RNAz calculates a normalized measure of thermodynamic stability by comparing the MFE m of a given (native) sequence to the MFEs of a large number of random sequences of the same length and base composition. A z-score is calculated as z = (m-μ)/σ,where μ and σ are the mean and standard deviations, resp., of the MFEs of the random samples. Negative z-scores indicate that a sequence is more stable than expected by chance. RNAz does not actually sample random sequences but approximates z-scores, which is much faster but of the same accuracy.
RNAz predicts a consensus secondary structure for an alignment by using the RNAalifold approach [3]. RNAalifold works almost exactly as single sequence folding algorithms (e.g. RNAfold), with the main difference that the energy model is augmented by covariance information. Compensatory mutations (e.g. a CG pair mutates to a UA pair) and consistentmutations (e.g. AU mutates to GU) give a "bonus" energy while inconsistent mutations (e.g. CG mutates to CA) yield a penalty. This results in a consensus MFE EA. RNAz compares this consensus MFE to the average MFE of the individual sequences <E> and calculates a structure conservation index: SCI = EA/<E>. The SCI will be high if the sequences fold together equally well as if folded individually. On the other hand, SCI will be low if no consensus fold can be found.
The two independent diagnostic features of structural ncRNAs, z-score and SCI, are finally used to classify an alignment as "structural RNA" or "other". For this purpose, RNAz uses a support vector machine (SVM) learning algorithm which is trained an a large test set of well known ncRNAs. RNAz reports a "RNA class probability" or P-value. Alignments with P>0.5 are classified as functional RNA. It is important to note that RNAz cannot distinguish functional RNA elements which are part of ncRNAs from elements which are cis-regulatory elements of mRNAs.
[1] Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs Proc. Natl. Acad. Sci. U.S.A. 102: 2454-2459, 2005
[2] Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, and Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem, 1994. 125:167-188.
[3] Hofacker IL, Fekete M, and Stadler PF. Secondary structure prediction for aligned RNA sequences. J Mol Biol, 2002. 319:1059-1066.
The web server operates in two different modes: In Standard Analysis mode, usually one single alignment is analyzed. In this mode, it is also possible to analyze more than one alignments in one session, but the alignments are treated to be independent from each other. In Genomic screen mode, a large number of alignments covering a genomic region can be screened and the results from all alignments are integrated in the end.
Multiple sequence alignments can be provided by cut-and-paste or uploaded as file. The server can currently read the following alignment formats: CLUSTALW, FASTA, PHYLIP,NEXUS, MAF, and XMFA. Alignments can be generated by any sequence based alignment program. However, one should not use "structurally enhanced" alignments generated by programs that consider RNA structures. Although this appears counter-intuitive, one has to keep in mind that RNAz was trained on pure sequence alignments and structural alignments could result in artifactually high scores even for alignments without conserved RNA structure.
To perform a screen of genomic regions in Genomic Screen mode your alignments need to fulfill some requirements: The identifier of the first sequence in the first alignment is used as reference. Each provided alignment must contain a sequence with this identifier and at least for this reference sequence correct genomic positions must be provided in the alignment. The identifier can be of the form species.chrom. In that case each alignment is required to have a sequence from the reference species. Below examples of valid alignments in MAF and XMFA format are shown, with sacCer1 as reference that must be present in all other alignments.
##maf version=1
a score=119673.000000
s sacCer1.chr4 1352453 73 - 1531914 GCCTTGTTGGCGCAATCGGTAGCGCGTATGACTCTT...
s sacBay.contig_465 14962 73 - 57401 GCCTTGTTGGCGCAATCGGTAGCGCGTATGACTCTT...
s sacKlu.Contig1694 137 73 + 4878 GCCTTGTTGGCGCAATCGGTAGCGCGTATGACTCTT...
s sacCas.Contig128 258 73 + 663 GCTTCAGTAGCTCAGTCGGAAGAGCGTCAGTCTCAT...
>1:1352453-1352526 + sacCer1.chr4
1531914 GCCTTGTTGGCGCAATCGGTAGCGCGTATGACTCTT...
>2:14962-15035 - sacBay.contig_465
GCCTTGTTGGCGCAATCGGTAGCGCGTATGACTCTT...
>3:137-210 + sacKlu.Contig1694
GCCTTGTTGGCGCAATCGGTAGCGCGTATGACTCTT...
>4:258-331 + sacCas.Contig128
GCTTCAGTAGCTCAGTCGGAAGAGCGTCAGTCTCAT...
= score = 119673
Choosing preprocessing options
The RNAz algorithm works globally, i.e. the given alignment is scored as a whole. For long alignments (e.g alignment of a whole chromosome), this is neither computationally tractable nor biologically meaningful. Therefore, long alignments are scanned in overlapping windows. The window and step-size can be set by the user. By default, a window size of 120 and a step-size of 40 is used. This window size appears large enough to detect local secondary structures within long ncRNAs and, on the other hand, small enough to find short secondary structures without loosing the signal in a much too long window.
In addition to this step, alignments are filtered in various ways before they are analyzed with RNAz. In particular, automatically generated genomic alignments are full of gap-rich regions, dubious aligned fragments, or low complexity regions. Such alignments are unlikely to contain true conserved structures and, in some cases, can cause artifactual predictions. Sequences that contain for example too many gaps or too many repeat-masked letters are therefore filtered out.
The RNAz program in its current implementation can only analyze alignments with up to six sequences. Six sequences usually hold enough information to allow reasonable predictions. If there are more sequences in the given alignment, the server selects an optimal subset of sequences. A greedy algorithm is used that gradually selects sequences to optimize for a given target diversity in the alignment. By default, a subset of six sequences is chosen which is optimized for a mean pairwise sequence identity of 80%.
At this stage, the user can choose different types of outputs to be generated. It is recommended to use the "Generate web-site" option since this output provides the best overview of the results (see below). For long jobs it is also recommended to provide an e-mail address to which a notification will be sent upon completion of the job.
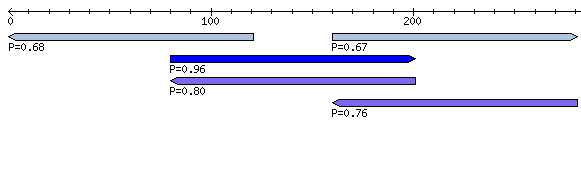
In Standard Analysis mode, an overview of each uploaded alignment is shown. Windows containing predicted secondary structures are highlighted and detailed information (z-score, structure conservation index, RNAz P-value, etc.) is shown in a table (see below). Arrows pointing to the right indicate forward reading direction relative to the uploaded alignment, while arrows pointing to the left indicate the reverse complement.
Results of an indivdual window
| Location | 80 - 200 | In Standard analysis mode this is the location of the window in the alignment. For genomic screens, the genomic location will be displayed. |
| Length | 120 | Length of the window (including gap columns). |
| Sequences | 6 | Number of sequences in the alignment. |
| Columns | 117 | Number of alignment columns (essentially Length without gap-only columns). |
| Reading direction | forward | "Forward" means in the same direction as given in the uploaded alignment. "Reverse" means the reverse complement of the uploaded alignment. |
| Mean pairwise identity | 80.69 | Average pairwise identity of the sequences in the alignment. |
| Mean single sequence MFE | -36.27 | Average minimum free energy of the individual sequences folded by RNAfold. |
| Consensus MFE | -31.53 | Consensus minimum free energy as calculated by the RNAalifold algorithm. It can be split into two terms as described below. |
| Energy contribution | -30.48 | The first term of the consensus MFE, which is the average folding energy from the standard energy model. |
| Covariance contribution | -1.05 | This is the second term of the consenus MFE and indicates "bonus" or "penalty" energies for compensatory/consistent and inconsistent mutations, respectively. |
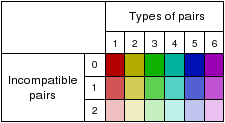
| Combinations/Pair | 1.28 | "Combinations/Pair" is a value that helps quantifying compensatory/consistent mutations. It is the number of different base pair combinations in the consensus structure divided by the overall number of pairs in the consensus structure. |
| Mean z-score | -2.64 | z-score calculated by RNAz. A z-score is calculated as z = (m-μ)/σ, where μ and σ are the mean and standard deviations, resp., of the MFEs of comparable random samples. Negative z-scores indicate that a sequence is more stable than expected by chance. |
| Structure conservation index | 0.87 | The SCI compares the consensus MFE EAderived by RNAalifold to the average MFE of the individual sequences <E> (SCI = EA/<E>). The SCI will be high if the sequences fold together equally well as if folded individually. |
| SVM decision value | 1.57 | Raw SVM score. For internal use only. It is converted to the more intuitive RNA-class probability (see below). |
| SVM RNA-class probability | 0.964815 | In alignments with P>0.5 a functional RNA is predicted. The higher this value, the more confident is the prediction. |
| Prediction | RNA | "RNA" if the SVM RNA-class probability is higher than 0.50, "other" otherwise. |
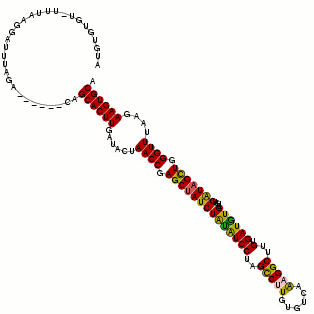
The predicted consensus secondary structure model is shown in various representations. Each representation uses the same coloring scheme for highlighting the mutational pattern with respect to the structure. If one predicted base-pair is formed by several different combinations of nucleotides consistent or compensatory mutations have taken place. This is indicated by different colors (see below). Pale colors indicate that a base-pair cannot be formed in some sequences of the alignment.
Secondary structure plot:
Structure annotated alignment. The alignment window is annotated with the predicted secodnary structure in dot/bracket notation and shaded according to the coloring scheme explained above. "."stands for an unpaired base, "(" corresponds to a paired base whose other base involved in the pair is further 3' and ")" corresponds to a paired base whose other base involved in the pair is further 5'.
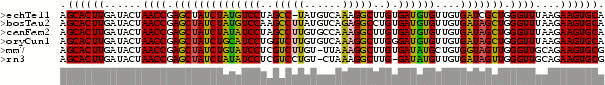
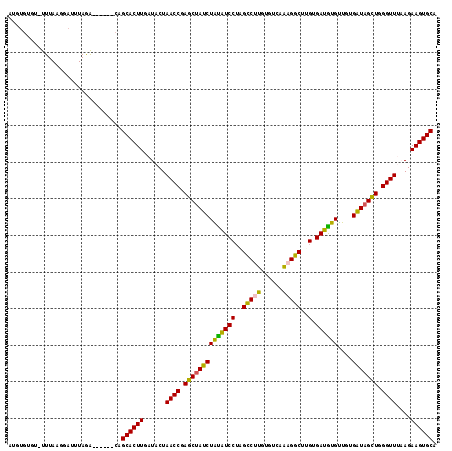
"Dot-Plot": The figure below shows an example of a "dot plot" of the
base-pairing matrix. In the upper half, the pairing-matrix of all bases are
shown with the pairing probability proportional to the squares shown. In
the lower half, shows the pairing matrix of the MFE structure with one
square for each predicted pair in this consensus structure.
The figure below shows the single sequences of the alignment in FASTA format together with their secondary structure in the dot/bracket notation and the MFE predicted by RNAfold. The last row is the consensus sequence together with the consensus structure and consensus MFE predicted by RNAalifold.

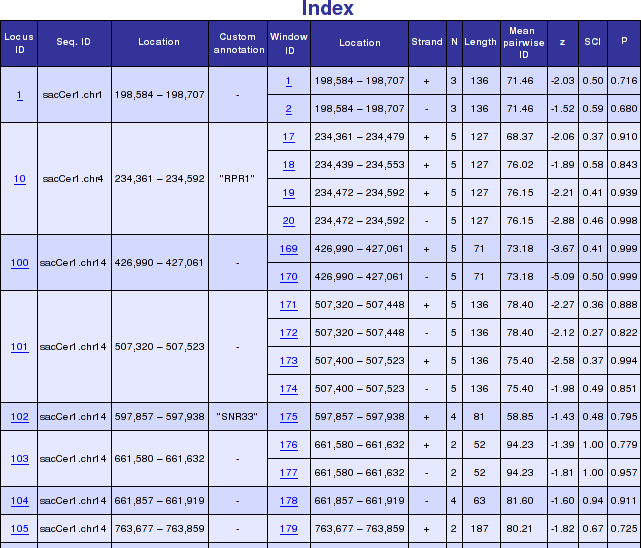
In Genomic Screen mode all overlapping windows with predicted RNA structures are combined to non-overlapping "loci". An overview table shows all these loci with their genomic location. In addition, a short overview of all windows contained within a locus is presented. More detailed informations and graphical representations as outlined above, can be accessed by following the hyperlinks. The figure below shows a typical genomic screen results with user uploaded custom annotation.