

| [Home|Help] |
Help topics
- General remarks
- Minimum free energy (MFE) structure
- Partition function folding and dot plots
- Ensemble free energy
- Centroid structure
- Circular folding
- Dangling ends
- Dot bracket notation
- Mountain plot
- Vienna RNA conservation coloring schema
- SVG files
- Postscript files
- Final Remarks
General remarks
Once the computation is complete you will be redirected to the results page, or you'll receive an email with the URL of the page. Note that the results will be deleted on our server after a while (currently 2 days). Download any files you want to keep to your local computer. The servers provide little protection for confidential data. Anyone who manages to guess the URL of your results page can access the data. Consider installing the software locally if this is an issue for you.
The Vienna RNA WebServers are based on the latest ViennaRNA Package (Version 2.6.3)
Minimum free energy (MFE) structure
The MFE structure of an RNA sequence is the secondary structure that contributes a minimum of free energy. This structure is predicted using a loop-based energy model and the dynamic programming algorithm introduced by Zuker et al. [1]. As an RNA secondary structure can be uniquely decomposed into loops and external bases the loop-based energy model treats the free energy F(s) of an RNA secondary structure s as the sum of the contributing free energies FL of the loops L contained in s. According to the chosen energy parameter set and a given temperature (defaults to 37 °C) the secondary structure s that minimizes F(s) is computed.
[1] Zuker, M. and Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information Nucleic Acid Res. 9(1): 133-148, 1981
Partition function folding and dot plots
In several bioinformatical applications one is not only interested in a single secondary structure with a special property like a minimum of free energy but in the probability of the occurance of a secondary structure si contained in the whole Boltzmann ensemble S. The partition function Q sums all Boltzmann weighted free energies of each secondary structure that is possible given an RNA sequence. This provides the possibility to calculate base pairing probabilities for each possible pair of bases. Therefore, the Vienna RNA Webservers utilize the algorithms implemented in the Vienna RNA Package [1] and output a base pairing probability matrix, the so called dot plot.
This dot plot consists of an upper and a lower triangle of a quadratic matrix. In both dimensions, each letter of the primary structure is assigned to a matrix index i and j, respectively. Matrix entries at position i,j are filled by black boxes indicating a base pair (i,j). In the upper triangle, the size of the boxes is proportinal to the base pairing probability where small boxes indicate low and large boxes high probability to form a base pair (i, j). The lower triangle is filled by boxes of equal size, depicting the secondary structure with minimal free energy. Considering alignments of RNA sequences results in colored dot-plots where the color represents sequence variation, as described in the Vienna RNA conservation coloring schema.

[1] McCaskill, J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure Biopolymers 29(6-7): 1105-1119, 1990
Ensemble free energy
If partition function folding was selected, an ensemble structure depicting the base pair probabilities summarized by pseudo bracket notation with the additional symbols ',', '|', '{', '}' is shown on the results page too. Here, the usual '(', ')', '.', represent bases with a strong preference (more than 2/3) to pair upstream (with a partner further 3'), pair down-stream, or not pair, respectively. '{', '}', and ',' are just weaker version of the above and '|' represents a base that is mostly paired but has pairing partners both upstream and downstream. In this case open and closed brackets need not match up. This pseudo bracket notation is followed by the ensemble free energy F = -kT ln Q in kcal/mol. Considering alignments of RNA sequences, these energies include the covariance term (they're not physical energies).
Centroid structure
The centroid structure of an RNA sequence is the secondary structure with minimal base pair distance to all other secondary structures in the Boltzmann ensemble.
Circular folding
Although most RNA molecules are linear, circular single stranded RNAs occur in a few cases. To take such circular RNAs into account the Vienna RNA Package implements additional memory efficient post- and pre-processing steps [1] for almost all included programs since Version 1.7 . If you use the Vienna RNA Webservers to treat circular RNAs check the option "assume RNA molecule to be circular" which is located in advanced options.
[1] Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures Bioinformatics 22(10): 1172-1176, 2006
Dangling ends
Additional stabilizing energies which arize if an unpaired nucleotide stacks with an adjacent base pair are called dangling-end contributions. When calculating the free energy of a loop, three possibilites for regarding dangling-end contributions exist. Firstly they may be ignored and therefore do not play a role in the algorithm. The second way is to take them into account for every combination of adjacent bases and base pairs. And thirdly, a more complicated energy model can be appliedletting unpaired bases stack with at most one base pair. The Vienna RNA Package implements each of these three models plus an additional energy contribution for coaxial stacking of helices. Using the Vienna RNA Webservers, you can switch dangling-end handling in advanced options.
Dot bracket notation
The bracket notation for RNA secondary structures Pseudo-knot free secondary structures can be represented in the space-efficient bracket notation, which is used throughout the Vienna RNA package. A structure on a sequence of length n is represented by a string of equal length consisting of matching brackets and dots. A base pair between base i and j is represented by a '(' at position i and a ')' at position j, unpaired bases are represented by dots. Thus the secondary structure
(((..((((...)))).)))is equivalent to:
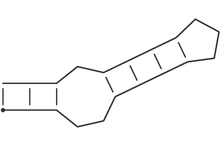
i.e. a stem-loop structure consisting of a an outer helix of 3 base pairs followed by an interior loop of size 3, a second helix of length 4, and a hairpin loop of size 3.
Mountain plot
A mountain plot represents a secondary structure in a plot of height versus position, where the height m(k) is given by the number of base pairs enclosing the base at position k. I.e. loops correspond to plateaus (hairpin loops are peaks), helices to slopes.

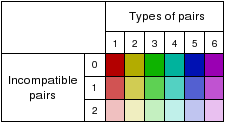
Vienna RNA conservation coloring schema
Representations that are generated by RNAalifold use a
coloring scheme for highlighting the mutational pattern
with respect to the structure. If one predicted base-pair
is formed by several different combinations of nucleotides
consistent or compensatory mutations have taken place. This
is indicated by different colors (see below). Pale colors
indicate that a base-pair cannot be formed in some
sequences of the alignment.
SVG files
A SVG output is produced by several severs, as this file format allows to easily generate interactive drawings. The web browser Firefox natively supports SVG without the need of installing additional plugins. If you are using the Internet Explorer please install the Adobe SVG plugin.
Postscript files
PostScript (PS) is a page description language (PDL) developed by Adobe. Each server will provide graphical output in form of PostScript files. These files can be used straight forward in professional type setting programs such as LaTeX. In addtion, we provide for each PS file a PDF file and a link to send the PS file to an online image converter that is able to export the file to several bitmap formats. Unfortunately, Microsoft Windows ships without a PostScript viewer. If your machine can't display postscript files install gsview.