

| [Home|Help] |
Help topics
Introduction
RNAstrand predicts the reading direction of a structured RNA gene from a multiple sequence alignment and its reverse complement. It is meant as a post processing tool for RNA prediction tools which in general do not reliable annotate the reading direction of the structured RNA candidate.
A simple way to determine the likely reading direction is to score an alignment and its reverse complement using RNAz or another tool for recognizing structured RNAs. However, one should use RNAstrand as it is more accurate. Direction information is needed e.g. to determine whether a conserved RNA motif is intronic, located within a coding sequence or an untranslated exon, an independent ncRNA, or one of the many classes of small RNAs associated with other transcripts.
Theory
Reading strand information is given in the small asymmetry in the energy rules and in particular by GU pairs that map to a non-canonical AC pair in the reverse complement of the alignment. Those asymmetries are utilized by four classification variables:
1) Difference in mean folding energies (meanMFE): Minimum free energies
of the secondary structures of single sequences are computed by RNAfold
from the Vienna RNA package.
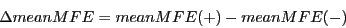
2) Difference in mean z-scores (meanZScore): z-scores of single sequences
are estimated by the same SVM-regression procedure as in RNAz.
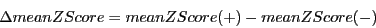
3) Difference in consensus minimum free energy (consMFE): Energy of
consensus secondary structure of the sequence alignment is computed
by RNAalifold from the Vienna RNA package.
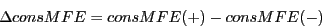
4) Difference in structure conservation index (SCI): SCI is defined as the ratio
of consensus energy and mean single sequence energy and quantifies to what
extent the single sequences fold into the same consensus structure.
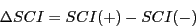
Descriptors 1 and 2 depict the energetic differences between both strands, while 3 and 4 capture the differences in structural conservation.
The significance of the strand differences are interpreted by:
5) Averaged mean pairwise identity (MPI) of both input alignments.
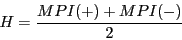
6) Number of sequences in input alignments.
n
7) Sum of fraction of GU base pairs in consensus secondary structure of both reading directions.
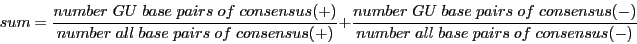
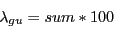
Descriptors 5, 6 and 7 provide information on the structure of the input alignments.
For detailed information on the SVM descriptors please read the main paper:
"RNAstrand: Reading Direction of Structured RNAs in Multiple Sequence Alignments"
Kristin Reiche and Peter F. Stadler, Algorithms for Molecular Biology, 2:6, 2007.
RNAstrand score
The RNAstrand score D is defined as
D = 2*P-1 (-1 <= D <= 1)
P is the SVM-class probability that RNA is in same reading direction than the input
alignment. D converges to +1 the larger the evidence that RNA is in reading direction of
input alignment and to -1 the larger the evidence that RNA is in reverse complement of
input alignment.
Strand is predicted according to a chosen threshold value c (default is 0.0001):
D > +c: RNA is in reading direction of input alignment
D < -c: RNA is reverse complement of input alignment
-c <= D <= +c: No decision of reading direction
Limitations
Due to characteristics of the training set no warranty about strand prediction is
given if the alignment is shorter than 40nt, longer than 400nt or contains more
than 6 sequences. Best classification results are yielded for alignments with
at least 60% mean pairwise sequence identity.
For long alignments we suggest to use a windowing technique to scan alignment
subregions independently.
In case a descriptor exceeds the range of the training data a warning message like
"WARNING: Descriptor 'delta mean z-score' is out of range (min=-4.9200 max=4.9200)."
is printed.
Contact
For any questions, comments to the software or bug reports please contact Kristin Reiche (kristin@bioinf.uni-leipzig.de).
Citation details
If you find this tool useful for your work please site it as follows: "RNAstrand: Reading Direction of Structured RNAs in Multiple Sequence Alignments" Kristin Reiche and Peter F. Stadler, Algorithms for Molecular Biology, 2:6, 2007.